
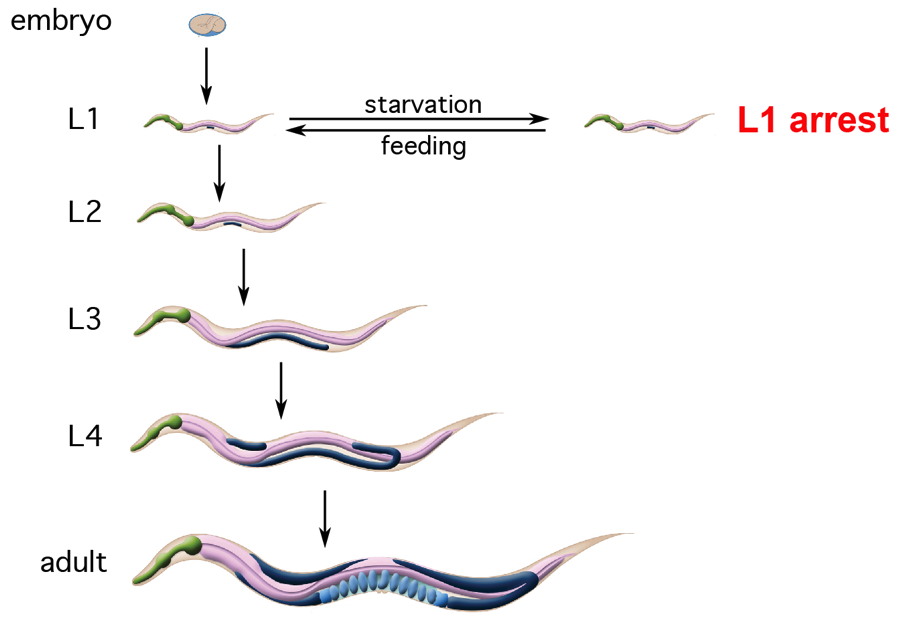
Life in the wild is feast or famine for C. elegans, and the worm has a variety of robust responses to starvation (reviewed in Baugh and Hu 2020). Larvae that hatch in the absence of food arrest development in the first larval stage (L1 arrest) and resume development in response to feeding (reviewed in Baugh 2013). We are interested in the signaling and gene regulatory mechanisms that enable development of an entire animal to stop and start in response to nutrient availability. We are also interested in the molecular mechanisms that enable arrested larvae to endure starvation. Despite the remarkable ability of worms to alternate between growth and quiescence, there are consequences to early-life starvation, including adult pathology as well as potentially adaptive multigenerational effects. We are using the worm as model to understand how early-life experience affects adult disease as well as transmission of nongenetic effects across generations.
Given their life history, short generation time, and ever-improving tools for genetic and genomic analysis, C. elegans is ideally suited for this research. This work is relevant to environmental adaptation and developmental robustness as well as cancer, aging, and metabolic disease.
Nutritional control of development

L1 arrest and recovery provide an opportunity to stop and start development in response to nutrient availability (reviewed in Baugh 2013). Understanding how developmental homeostasis is maintained with fluctuating nutrient availability is critical to understanding phenotypic robustness. For example, how is development coordinated across the animal in transition between growth and quiescence? We are using this system to investigate signaling and gene regulatory mechanisms that enable the worm to reversibly arrest development.
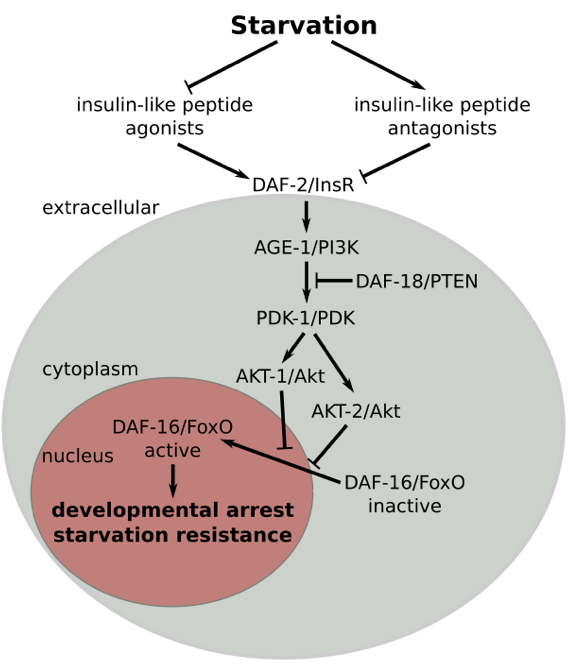
Insulin signaling regulates L1 arrest (Baugh 2006). Disruption of the insulin receptor daf-2 causes fed larvae to arrest development. Conversely, disruption of its transcriptional effector, the FoxO transcription factor daf-16, causes starved larvae to develop. However, daf-16/FoxO regulates development cell-nonautonomously by inhibiting TGF-ß and steroid hormone signaling pathways that otherwise promote development (Kaplan, Chen 2015). Intriguingly, the genome encodes 40 insulin ligands. Chemosensory neurons and intestinal cells both contribute to systemic regulation (Chen 2014, Kaplan 2018). daf-16/FoxO regulates transcription of the majority of the insulin genes, producing widespread positive and negative feedback regulation that affects developmental dynamics and homeostasis (Kaplan, Maxwell 2019). This complex signaling network coordinates growth, development and metabolism so that coherent physiological states are maintained across the animal as it responds to varying environmental conditions (Kaplan 2016).
L1 arrest and recovery provide an excellent opportunity to study nutritional control of gene expression. Several thousand genes are differentially expressed in fed and starved larvae, and starved larvae respond rapidly to feeding (Baugh 2009, Maxwell 2012). RNA Polymerase II is recruited to growth genes during L1 arrest where it remains inactive but poised until feeding (Maxwell 2014). Nutritional control of Pol II initiation and elongation contributes to rapid, widespread changes in gene expression in transition from quiescence to growth. Arrested larvae actively maintain a transcriptional response to starvation throughout the soma, but the germline remains in a transcriptional quiescent state during L1 arrest while germline transcripts display exceptional stability (Webster 2022a). Maintenance of germline transcriptional quiescence is essential to reproductive success upon recovery.
Genetic analysis of starvation resistance

L1 arrest provides a ripe opportunity to investigate the genetic basis of starvation resistance (reviewed in Baugh 2013 and Baugh and Hu 2020). Given the life history of C. elegans and related nematodes, starvation resistance is critical to fitness. Starvation resistance is also intimately related to aging and metabolic disease. Indeed, genes known to affect starvation resistance in C. elegans also influence aging throughout metazoa and are associated with diabetes in humans.

As in humans, early-life starvation in worms has pathological consequences later in life. Extended L1 starvation reduces reproductive success in adults (Jobson, Jordan 2015) by causing the development of germline tumors and other abnormalities in reproductive development (Jordan 2019). Reduction of insulin/IGF signaling during recovery from L1 arrest reduces starvation-induced gonad abnormalities, suggesting hyperactive insulin/IGF signaling in well fed larvae that were previously starved. We found that early-life starvation and insulin/IGF signaling affect expression of thousands of genes in adults, including numerous genes involved in lipid metabolism and Wnt signaling. Knock-down of lipid metabolism or Wnt signaling during recovery from L1 arrest suppresses starvation-induced gonad abnormalities as well (Jordan 2022; Shaul 2022). We are investigating the insulin/IGF signaling, lipid metabolism, and Wnt signaling network and how it affects development of germline tumors and other developmental abnormalities following early-life starvation.
Insulin signaling regulates starvation resistance (Baugh 2006). Disruption of the insulin receptor daf-2 increases resistance, and disruption of its transcriptional effector, the FoxO transcription factor daf-16, decreases resistance. daf-16/FoxO orchestrates a metabolic shift during starvation, promoting gluconeogenesis and synthesis of the disaccharide trehalose to maintain survival (Hibshman 2017). We are investigating additional effector mechanisms by which daf-16/FoxO promotes starvation resistance as well as daf-16-independent effects of insulin signaling on resistance. We are also interested in other pathways that affect starvation resistance.

We are developing innovative approaches to unbiased genetic analysis of starvation resistance that leverage the awesome power of sequencing. These approaches are based on selection of genetically diverse populations for starvation resistance and use of sequencing to measure changes in strain/allele frequency. To study natural variation, we pool wild strains to create the experimental population, use sequencing to assess relative starvation resistance of each strain, genome-wide association analysis to identify quantitative trait loci (Webster 2019), and fine mapping and genome editing to identify specific genetic variants. We identified four members of a single gene family that affect starvation resistance in wild strains of C. elegans , and we found that they do so by modifying insulin/IGF signaling (Webster 2022). We are also using mutagenesis to create experimental populations for analysis by population selection and sequencing. By saturating the genome with mutations, this approach should allow us to identify all genes that affect starvation resistance.
Multigenerational adaptation to starvation

With a short generation time and robust epigenetic mechanisms, C. elegans is an ideal model to study epigenetic inheritance. There is increasing evidence that epigenetic effects are transmitted across generations in this and other animals, but the extent to which they mediate environmental adaptation is unclear (Baugh and Day 2020). Consequently, it is difficult to assess the ecological or evolutionary significance of epigenetic inheritance. Furthermore, mechanisms of epigenetic inheritance are still being discovered, with the potential to make fundamental contributions to our understanding of phenotypic variation and disease risk in humans.

Dietary restriction causes worms to produce fewer but larger progeny (Hibshman 2016). These progeny are also more resistant to L1 starvation upon hatching. Dietary restriction alters insulin signaling to increase maternal oocyte provisioning of vitellogenin yolk protein (Jordan 2019). Increased yolk provisioning protects progeny from the pathological consequences of starvation by influencing insulin signaling in progeny. This work shows that insulin signaling in mother and offspring mediates adaptation to nutrient stress across generations. Furthermore, it identifies oocyte provisioning as an epigenetic mechanism and yolk protein as an epigenetic factor. To further elucidate this fascinating example of epigenetic inheritance, we are investigating how oocyte provisioning is regulated and how yolk affects insulin signaling in progeny.
Early-life starvation increases starvation resistance transgenerationally. Though L1 starvation reduces reproductive success in that generation, it increases heat resistance three generations later and starvation resistance at least two generations later (Jobson, Jordan 2015). Likewise, extended starvation during dauer diapause (a developmentally arrested alternative third-larval stage) increases starvation resistance and lifespan three generations later while affecting expression of nutrient-responsive genes (Webster 2018). These results suggest that transgenerational epigenetic inheritance could mediate environmental adaptation. This system provides an opportunity to identify potentially adaptive mechanisms for germline epigenetic memory.